Spinal Müsküler Atrofi (SMA) nedir, nasıl bir hastalıktır? Spinal Müsküler Atrofi (SMA) hastalığı tipleri, belirtileri, yaşam süreleri ve tedavisi.
Spinal Müsküler Atrofi (SMA)
Spinal Müsküler Atrofi nedir?
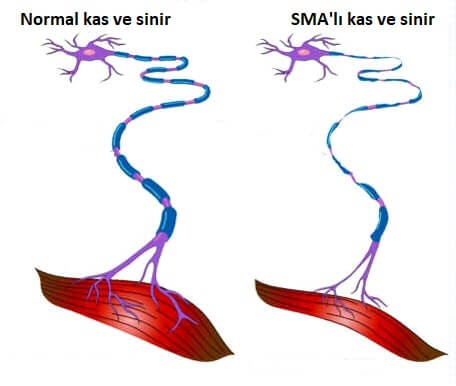
Spinal musküler atrofi (SMA), beyin sapı ve omurilikteki konuşma, yürüme, nefes alma ve yutma gibi temel kas aktivitesini kontrol eden çeşitli motor nöronların, giderek yok olduğu kalıtsal hastalıklardan biridir. Alt motor nöronlar kollardaki, bacaklardaki, göğüsteki, boğazdaki hareketleri kontrol eder.
Alt motor nöronlar ve kaslar arasındaki sinyallerde bozulmalar olduğunda, kaslar giderek zayıflar ve boşa harekete başlar ve kontrolsüz seğirme (fasikülasyonlar) oluşur. Üst motor nöronlar (beynin içinde bulunur) ile alt motor nöronlar arasındaki sinyallerde bozulmalar olduğunda, ekstremite kaslarda sertlik (spastisite olarak adlandırılır) gelişir, hareketler yavaş ve efor gerektirir hale gelir ve diz ve ayak bileği gibi tendon refleksleri aşırı aktif hale gelir. Zamanla, hareketi kontrol etme olanağı kaybolabilir.
Spinal Müsküler Atrofi neden olur?
Spinal Müsküler Atrofi, motor nöronların (SMN proteini) hayatta kalması için önemli olan bir proteini üreten SMN1 genindeki kusurlardan kaynaklanır. Spinal Müsküler Atrofide SMN proteininin yetersiz seviyeleri alttaki motor nöronların dejenerasyonuna, iskelet kası güçsüzlüğüne neden olur. Bu zayıflık gövde ve üst bacak ve kol kaslarında, el ve ayaklardaki kaslardan daha sıktır.
Spinal musküler atrofi kalıtsal mıdır?
Çocuklarda Spinal Müsküler Atrofi bozuklukları anotozomal resesif bir tarzda kalıtsaldır. Otozomal resesif geçiş, çocuğun her iki ebeveynden de kusurlu genin bir kopyasını olması gerektiği anlamına gelir. Bu ebeveynlerde hastalık belirtileri olmasını gerektirmeyen bir durumdur. Otozomal resesif hastalıklar genellikle aynı nesilde (kardeşler veya kuzenler) birden fazla kişiyi etkiler.
Kennedy hastalığı, hastalığın yetişkinler için X kromozomuna bağlı kalıtsal bir formudur; bu, hastanın annesinin kusurlu geni X kromozomlarından birini taşıdığı ve rahatsızlığı oğullarına ilettiği anlamına gelir. Erkekler, annelerinden bir X kromozomu ve babalarından bir Y kromozomu, kadınlar her bir ebeveynden bir X kromozomu alırlar. Kız çocuklarının, annelerinin hatalı X kromozomunu ve babalarından güvenli bir X kromozomu miras alma şansı yüzde 50’dir; bu da onlara mutasyona karşı asemptomatik taşıyıcılar yaparlar.
 Spinal musküler atrofi tipleri ve belirtileri nelerdir?
Spinal musküler atrofi tipleri ve belirtileri nelerdir?
Çocuklarda Spinal musküler atrofi, başlangıç yaşlarına, şiddetine ve semptomların ilerlemesine göre üç tipe ayrılır. Her üç tip de SMN1 genindeki kusurlardan kaynaklanmaktadır.
Werdnig-Hoffmann hastalığı olarak da adlandırılan spinal kas atrofisi tip 1, çocuğun 6 aylık olduğu zamana kadar belirgindir. Semptomlar hipotoni (kas gücünün azalması), ekstremite hareketlerinin azalması, tendon refleksleri eksikliği, fasikülasyonlar, titreme, yutma ve beslenme güçlüğü ve solunum bozukluğunu içerebilir. Bazı çocuklar ayrıca skolyoz (omurganın eğriliği) veya diğer iskelet bozuklukları geliştirir. Etkilenen çocuklar oturamaz ya da ayakta kalamazlar ve büyük çoğunluğu genellikle 2 yaşından önce solunum yetmezliğinden ölürler. Bununla birlikte, Spinal musküler atrofi tip I olan bireylerde sağkalım oranı, son yıllarda artmıştır.
Ara form olan Spinal musküler atrofi tip II‘nin semptomları genellikle 6-18 aylık dönemlerde başlar. Çocuklar destek olmadan oturabilirler ancak ayakta durmakta ya da yürümekte güçlük çekerler ve solunum yolu enfeksiyonu riski artışı da dahil olmak üzere solunum zorlukları yaşayabilirler. Hastalığın ilerlemesi değişkenlik göstermektedir. Yaşam süresi kısadır, ancak bazı kişiler ergenliğe veya genç erişkinliğe kadar yaşarlar.
Spinal musküler atrofi tip III (Kugelberg-Welander hastalığı) belirtileri 2 ila 17 yaş arasında görülür ve anormal yürüyüş; Koşu zorluğu, merdiven çıkma zorluğu ve parmaklarda hafif titreme görülür. Alt ekstremite sıklıkla etkilenir. Komplikasyonlar arasında skolyoz ve eklem kontraktürleri-eklemlerin serbestçe hareket etmesini önleyen, anormal kas güçsüzlüğü ve zayıflığından kaynaklanan kasların veya tendonların kronik kısalması bulunur. Spinal musküler atrofi tip III’lü kişiler solunum yolu enfeksiyonlarına eğilimli olabilir, ancak normal bir ömür süresine sahip olurlar.
Kennedy hastalığı, çocukluk yaşlarında görülmez, ilk önce 15 ila 60 yaş arasında fark edilir. Belirtilerin başlangıcı değişir ve yüz, çene ve dil kaslarının zayıflığı ve atrofisini içerir ve çiğneme, yutma ve konuşmada değişiklikler ile sonuçlanır. Erken belirtiler kas ağrısı ve yorgunluk içerebilir. Vücudun gövdeye en yakın kol ve bacak kaslarında zayıflık, kas atrofisi ve fasikülasyonlar ile birlikte zamanla gelişir. Kennedy hastalığı olan bireyler de ayaklarda ve ellerde his kaybı oluşur. Sinir iletim çalışmaları neredeyse tüm bireylerin duyu nöropatisine (duyusal sinir iltihabından veya dejenerasyonundan kaynaklanan ağrı) sahip olduklarını doğrulamaktadır. Etkilenen erkek bireylerin göğüslerinde büyüme olabilir veya insüline bağımlı diabetes mellitus gelişebilir.
Spinal musküler atrofi nasıl teşhis edilir?
SMN1 genindeki hataların veya mutasyonların olup olmadığını gösterecek bir test mevcuttur. Bu test Spinal musküler atrofi Tip I, II ve III’ün en az % 95’ini teşhis eder. Diğer diagnostik testler, beyin ve / veya omurilikten gelen elektriksel aktiviteyi, sinir iletim hızını, sinyal gönderme yeteneğini değerlendiren elektromiyografi, kas biyopsisi (nöromüsküler bozuklukları teşhis etmek için kullanılır ve bir kişinin kusurlu bir genin taşıyıcısı olup olmadığını belirler), kan ve idrar testidir.
Spinal musküler atrofinin tedavisi var mı?
Spinal musküler atrofi tedavisi yoktur. Tedavi, semptomların yönetimi ve komplikasyonların önlenmesinden oluşur.
Kas gevşeticiler spastisiteyi azaltabilir. Aşırı tükürük üretimi kontrol altına alınabilir. Antidepresanlar depresyon tedavisinde faydalı olabilir.
Fizik tedavi, mesleki terapi ve rehabilitasyon duruşu düzeltmeye, eklem hareketsizliğini önlemeye ve kas güçsüzlüğünü ve atrofisini yavaşlatmaya yardımcı olabilir. Germe ve güçlendirme egzersizleri spastisitenin azalmasına, hareket aralığının yükselmesine ve dolaşımı arttırmaya yardımcı olabilir. Bazı kişilere konuşma, çiğneme ve yutkunma güçlükleri için ilave terapi gerektirir. Isı uygulamak kas ağrısını hafifletebilir. Destekler, ortotikler, konuşma sentezleyicileri ve tekerlekli sandalyeler gibi yardımcı cihazlar, bazı kişilerin bağımsızlığını korumasına yardımcı olabilir.
Dengeli ve düzenli beslenme, ağırlık ve dayanıklılığın korunması için gereklidir. Çiğneyemeyen ya da yutamayan kişilere besleme tüpünün takılmasını gerektirebilir. Gece non-invazif ventilasyon, uyku apnesini önleyebilir ve bazı kişilerde gündüz boyun, boğaz ve göğüste kas güçsüzlüğü nedeniyle nefes almayı kolaylaştırmak için solunum cihazı gerekebilir.
Spinal kas atrofisi olan bir hasta ne kadar yaşar?
Spinal kas atrofisinin bazı biçimleri ölümcüldür.
Kennedy hastalığının seyri değişir, ancak genellikle yavaş yavaş ilerlemektedir. Bireyler hastalığın ilerleyen dönemlerine kadar ayakta kalabilirler. Kennedy hastalığı olan bireylerin yaşam beklentisi genellikle normaldir.
Spinal kas atrofisi olan insanlar uzun süre stabil kalabilirler ancak iyileşme beklenmemelidir.
![]()
